

Research Interests of the Straub Group
pdf version (better image quality)
Our research covers several topics of organometallic chemistry.
-
Investigation of the mechanism of the copper-catalyzed azide alkyne cycloaddition reaction (Huisgen Sharpless triazole click reaction), particularly the origin of the second-order dependence of the reaction rate on the concentration of copper(I) ions. [1]
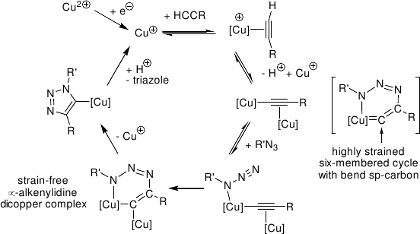
We isolated a copper(I) triazolide complex,[2] which has hitherto only been postulated as an intermediate.
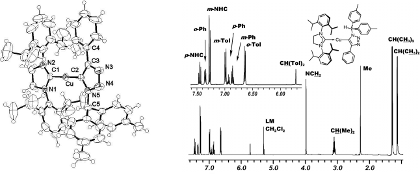
The triazolide complex is formed at r.t. from an organoazide and an acetylide complex. The triazolide is thermally stable in the solid state and in solution, but it is rapidly protonated by e.g. acetic acid. Apparently, N2 elimination from the triazolide ligand does not occur with the substitution pattern present in this click intermediate.
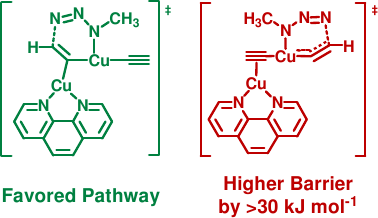
In a quantum-chemical model study, we identified thermodynamically stable dinuclear and tetranuclear copper acetylide structures as reasonable starting catalysts.[1] Mononuclear model species and aqua complexes have to be considered as irrelevant, high-energy species. Dinuclear copper(I) complexes with bridging acetylides are predicted to be both thermodynamically more stable and more reactive from a kinetic perspective. The favored (green) pathway comprises two copper atoms binding to an evolving sp² carbon in the rate-determining step. In the disfavored (red) alternative, a highly ring-strained sp-type carbon in a six-membered cycle would be enforced, thereby increasing the formation barrier by an additional >30 kJ mol-1.
Additives such as phenanthroline are proposed to increase the efficiency of CuAAC click reactions by – among other reasons – inhibiting aggregation of the copper acetylides.
[1] B. F. Straub*, Chem. Commun. 2007, 3868.
[2] C. Nolte, P. Mayer, B. F. Straub*, Angew. Chem. 2007, 119, 2147; Angew. Chem. Int. Ed. 2007, 46, 2101.
-
Synthesis of non-homoleptic tertiary phosphines, tailoring ligands for transition metal catalysis in organic synthesis. A didactic and aesthetic phosphine synthesis is the preparation of the “3-2-1 phosphine” depicted below.[3]
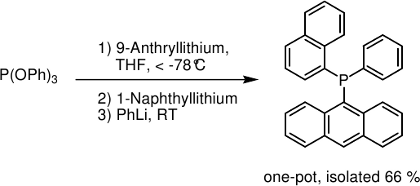
[3] J. Keller, C. Schlierf, C. Nolte, P. Mayer, B. F. Straub, Synthesis 2006, (2), 354.
-
We develop novel coinage metal catalysts inspired by the ethene receptor protein ETR1.[4] The latter is responsible for the recognition of traces of the plant hormone ethene by plants, leading to e.g. stress responses or fruit ripening. The core of ETR1 is an uncharged copper(I) imidazole thiolate unit, stabilized against cluster formation by steric shielding due to the protein backbone.
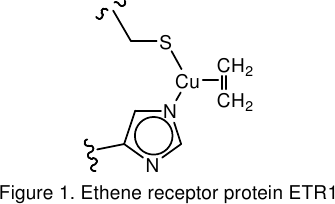
We tailor chelating, electron-rich monoanionic thiolate ligands with sterically demanding substituents to mimic the electronic and steric situation of the active protein site.
[4] a) F. I. Rodríguez, J. J. Esch, A. E. Hall, B. M. Binder, G. E. Schaller, A. B. Bleecker, Science 1999, 283, 996;
b) A. B. Bleecker, H. Kende, Annu. Rev. Cell. Dev. Biol. 2000, 16, 1.
-
We investigate mechanisms of transition metal catalyzed reactions by DFT model calculations. Convential experimental methods are often not suited to gain information about reactive intermediates or transition states that are neither the rate- nor the selectivity-determining step.
-
Palladium-catalyzed cyclopropanation of alkenes by CH2N2
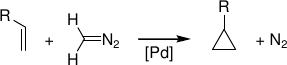
Predicted mechanism:
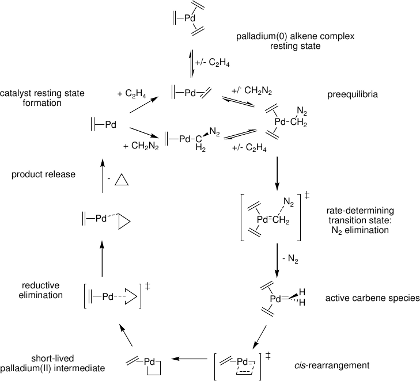
[5] B. F. Straub, J. Am. Chem. Soc. 2002, 124, 14195.
-
Reppe’s Nickel-catalyzed Ethyne Tetramerization
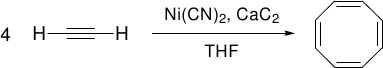
Predicted mechanism:
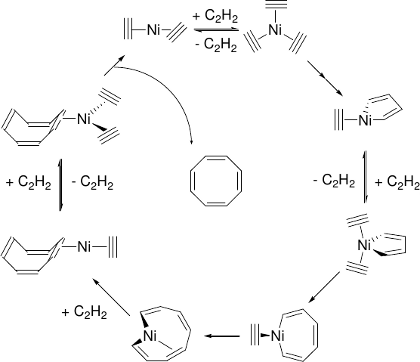
[6] W. Reppe, O. Schlichting, K. Klager, T. Toepel, Justus Liebigs Ann. Chem. 1948, 560, 1.
[7] B. F. Straub, C. Gollub, Chem. Eur. J. 2004, 10, 3081.
-
Gold-catalyzed Benzannulation
In Y. Yamamoto’s benzannulation reaction,[8] it remains unclear whether gold(III) or reduced gold(I) species are the active catalyst.
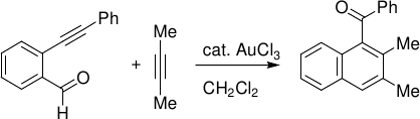
In our DFT study,[9] both AuCl and AuCl3 are predicted to feature almost identical overall Gibbs free activation energies. Surprisingly, no direct [4+2] cycloaddition step can be located, but a pathway comprising a [3+2] cycloaddition with subsequent rearrangements.
Predicted mechanism (X = 1 or X = 3):
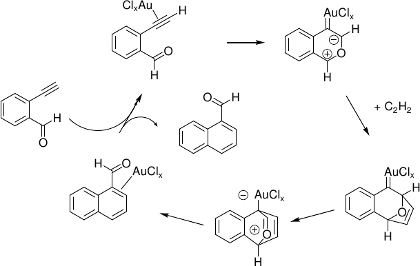
[8] (a) N. Asao, K. Takahashi, S. Lee, T. Kasahara, Y. Yamamoto, J. Am. Chem. Soc. 2002, 124, 12650. (b) N. Asao, T. Nogami, S. Lee, Y. Yamamoto, J. Am. Chem. Soc. 2003, 125, 10921. (c) N. Asao, K. Sato, Y. Yamamoto, Tetrahedron Lett. 2003, 44, 5675. (d) G. Dyker, D. Hildebrandt, J. Liu, K. Merz, Angew. Chem. 2003, 115, 4736; Angew. Chem. Int. Ed. 2003, 42, 4399.
[9] B. F. Straub, Chem. Commun. 2004, 1726.
-
The origin of the differences of metathesis activity of ruthenium carbene complexes (first generation versus second generation Grubbs catalysts) is due to the electronic and steric stabilization of active and inactive carbene conformations.[10] Strongly electron-donating spectator ligands L such as N-heterocyclic carbenes stabilize the active carbene conformation and facilitate the [2+2] cycloaddition to a ruthenacyclobutane intermediate. Alkene ligand rotation in the alkene carbene complexes is rapid and the alkene ligand conformations are almost degenerate.
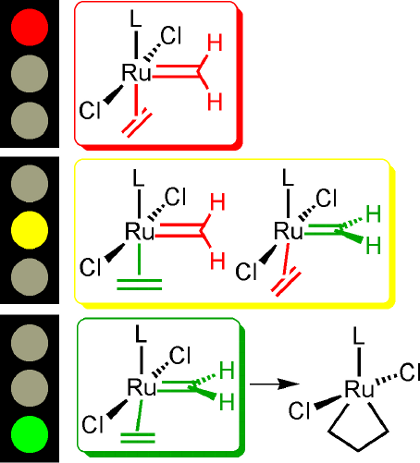
[10] (a) B. F. Straub, Angew. Chem. 2005, 117, 6129; Angew. Chem. Int. Ed. 2005, 44, 5974; (b) B. F. Straub*, Adv. Synth. Catal. 2007, 349, 204.
-
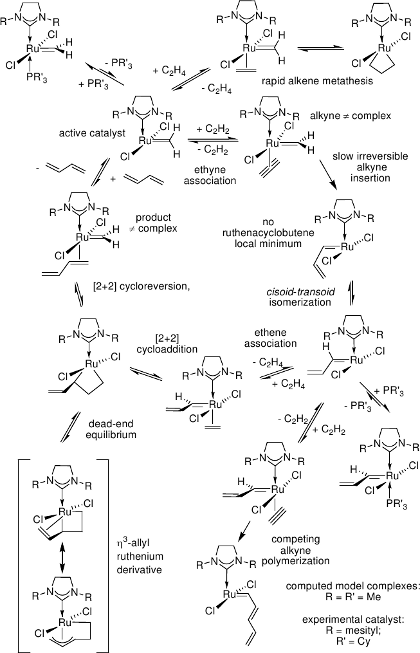
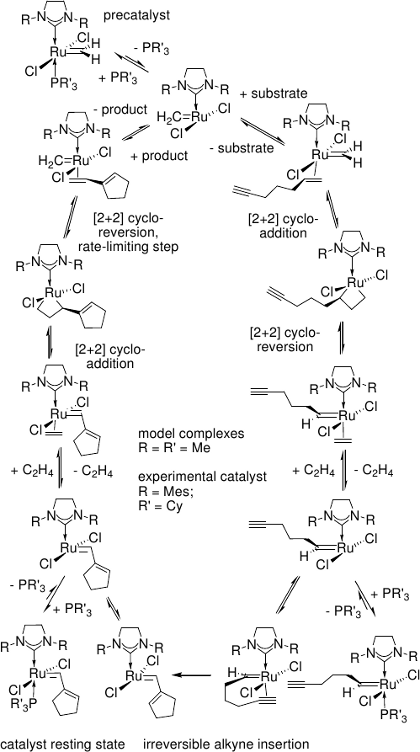
In ruthenium-carbene catalyzed enyne metathesis,[11] ruthenacyclobutenes are not intermediates in the catalytic cycle, but only transient structures with the lifetime of a molecular vibration.[12]
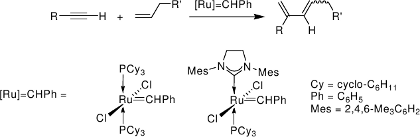
All relevant steps of the catalytic cycle have been modelled for both the intra- and intermolecular reaction of alkene and alkyne substrate.
[11] (a) S. T. Diver, A. J. Giessert, Chem. Rev. 2004, 104, 1317; (b) S. T. Diver, A. J. Giessert, Synthesis 2004, 466; (c) C. S. Poulsen, R. Madsen, Synthesis 2003, 1; (d) M. Mori, Top. Organomet. Chem. 1998, 1, 133.
[12] J. J. Lippstreu, B. F. Straub, J. Am. Chem. Soc. 2005, 127, 7444.
-
Palladium-catalyzed cyclopropanation of alkenes by CH2N2
pdf version (better image quality)
Seitenbearbeiter:
E-Mail
Letzte Änderung:
22.06.2014