
Current Research
Identification of epigenetically silenced tumor suppressor gene candidates in glioblastomas. (Dr. W. Mueller)
Epigenetic silencing of genes by promoter hypermethylation constitutes an alternative way of gene inactivation. In an effort to identify novel tumor suppressor gene candidates (TSG), we took advantage of the reversibility of epigenetic silencing by pharmacological manipulation of cultured gliomas with the substance 5`-aza 2`-deoxicitidine (5`-aza-dC). Combined with a genome wide, microarraybased gene expression profiling it was possible to identify novel TSG candidates, that were inactivated primarily by promoter hypermethylation. The expression profile of cells that were not treated with 5`-aza-dC where compared to the expression profile of the same cells following demethylation treatment. Genes that reveal significant up-regulation of their expression after 5`-aza-dC manipulation are candidates to be tested in ongoing studies. Methylation patterns in promoters of RUNX3 and TES have already been determined (Figure). Candidates with differential methylation in glioblastomas may proof to be relevant prognostic or predictive markers such as MGMT or future targets for tailored therapy.
Molecular diagnostics in phase I, phase II and phase III trials. (PD Dr. C. Hartmann)
We provide molecular diagnostics for the following studies: RTC 22033-26033 (Evaluating primary chemotherapy with temozolomide vs. radiotherapy in patients with low grade gliomas with stratification for genetic 1p/19q loss: a phase III study) is coordinated by the European Organization for Research and Treatment of Cancer (EORTC). The objectives are to compare the progression-free survival of patients with low-grade gliomas treated with radiotherapy versus temozolomide andto compare overall survival, the incidence of late toxicity, toxic effects and the quality of life of patients treated with these regimens. Patients are stratified according to participating centre, chromosome 1p status, contrast enhancement on MRI, age, and WHO performance status. Patients are randomized to 1 of 2 treatment arms. Lilly LY317615 (Enzastaurin (LY317615) with concomitant radiation therapy, followed by enzastaurin maintenance therapy in subjects with newly diagnosed glioblastoma - a multicenter, open-label, uncontrolled Phase II study) is coordinated by the Klinische Kooperationseinheit Neuroonkologie (DKFZ). The objectives are evaluation of progression-free survival in patients with newly diagnosed glioblastoma without promoter methylation of the MGMT gene treated with enzastaurin and to investigate safety and tolerability. NOA-04 (Randomized phase III trial of sequential radiochemotherapy for anaplastic oligodendroglial and astrocytic gliomas WHO grade III using PCV or temozolomide) is coordinated by the department for neurology at the university Tübingen. The objectives are to determine time to second progression or time to death before second progression in patients treated either by Temozolomide or PCV chemotherapy before radiation therapy or Temozolomide or PCV chemotherapy after radiation therapy and to validate differences in outcome of patients with anaplastic oligodendroglial and astrocytic gliomas in respect to 1p/19q status and MGMT methylation status. UKT-05 (Radiotherapy and concomitant low-dose Indometacin and temozolomide therapy and adjuvant temozolomide therapy (one week on/one week off) in newly diagnosed glioblastoma: a phase II study) is coordinated by the department for neurology at the university Tübingen. The objective is to test if patients treated by intensified temozolomide chemotherapy in conjunction with the COX inhibitor indometacin during radiation therapy followed by weekly alternating temozolomide chemotherapy perform better than those patient in the chemotherapeutical arm of the EORTC 26981 study. GGN-II/CP1 (German Glioma Network/Central Project 1) is coordinated by the Klinische Kooperationseinheit Neuropathology (DKFZ) Microarray-based profiling will be performed at the genomic and transcriptional level to systematically characterize molecular aberrations in glioblastomas.
Neurofibromatosis type I (D. Reuss)
Plexiform neurofibromas in NF1 patients are at risk for transformation to malignant peripheral nerve sheath tumors (MPNST). It is widely agreed on that the occurrence of an additional somatic mutation within the second copy of the NF1 gene in a Schwann cell population is an essential early step in the pathogenesis of both neurofibroma and MPNST. The only well-characterized function of the NF1 gene product neurofibromin is its RasGAP activity, contained in the central GAP related domain. Neurofibromin function also seems to be involved in the cAMP protein kinase A (PKA-) pathway. We aim at identification and characterisation of new Ras-dependent and Ras-independent functions of neurofibromin. Furthermore we focus on the identification of potential new therapeutic targets to target Schwann cells harbouring a somatic NF1 mutation thus allowing a therapy that does not exhibit substantial toxic side effects on other cell types. As new tumors arise throughout the life of patients with NF1, we are also interested in evaluating mechanisms of tumor immune escape in peripheral nerve sheath tumors and their implications for pathogenesis and therapeutic approaches.
Apoptosis in malignant gliomas (Dr. M. Siegelin)
Despite aggressive multimodal treatment, malignant gliomas are almost always fatal. The resistance to apoptosis contributes to low radiation and drug sensitivity of glioma cells. The presence of high levels of anti-apoptotic factors, such as the inhibitor of apoptosis proteins (IAP) confers resistance to cancer cells. One of our interests focuses on IAP- Proteins and their antagonists. For this purpose, we analyze protein interactions between hypoxia-induced proteins and IAP proteins. Special attention is given to migration, proliferation and apoptosis of hypoxic glioma cells in respect to modifications in IAPs.
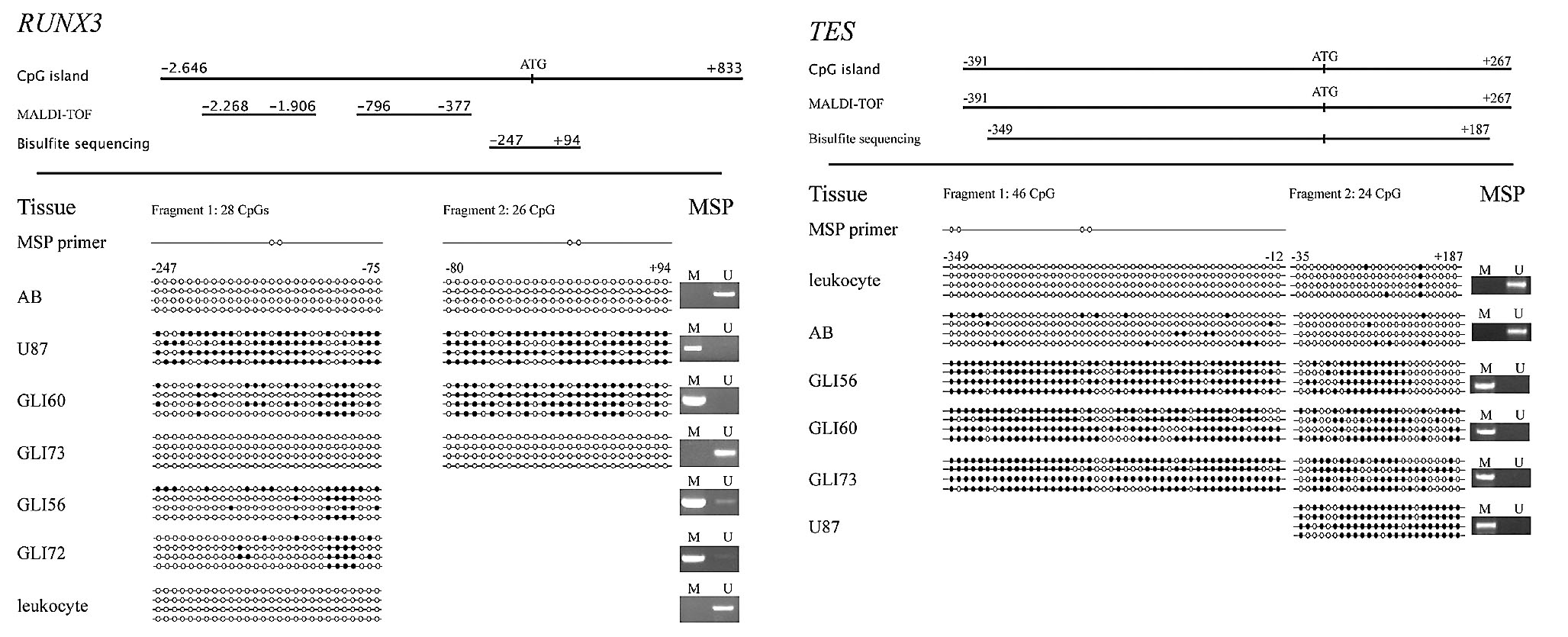 |
| Fig. 1: Bisulfite sequencing and MSP (methylation sensitive PCR ) of RUNX3 and TES in primary cultures of glioma, U87 and controls. Presence or absence of methylated CpGs matches the results of MSP. Black dot - methylated CpG; white dot - unmethylated CpG; M - methylated in MSP assay; U - unmethylated in MSP assay |