
Current Research
Neurotransmitter release is the paradigm for regulated exocytosis and distinct components acting at different steps have been identified (see Figure 1 for a brief overview). Membrane fusion is initiated by the interaction of the v-SNARE VAMP2 on synaptic vesicles with its cognate t-SNARE, consisting of syntaxin1 and SNAP25 on the presynaptic plasma membrane. SNARE proteins assemble into a stable four-helix bundle. In vitro assays have shown that this protein folding reaction drives membrane fusion. However, in vivo, SNARE complex assembly is tightly regulated and involves multiple steps and numerous regulatory components that either accelerate or arrest the formation of distinct reaction intermediates. The initial tethering of synaptic vesicles to the active zone of the neuronal synapse does not depend on v-/t-SNARE interactions, but likely requires small GTPbinding proteins such as Rab3 and its effectors. This vesicle tethering machinery is directly linked to the downstream fusion machinery. A multi-step, multi-component process controls SNARE complex assembly. The majority of the synaptic vesicles seems to be docked by partially assembled SNARE complexes and might be arrested at a hemifusion step. Calcium binding to the calcium sensing machinery, which is coupled to the SNAREs, opens the fusion pore. Fusion pores have two fates: they can reversibly close or fully dilate resulting in the complete incorporation of the vesicular components into the plasma membrane. SNAREs are subsequently recycled by an energy consuming reaction, involving the hexameric ATPase NSF. Endocytosis regenerates the synaptic vesicles.
To analyze the dynamic assembly of the neuronal exocytosis machinery and to dissect the distinct molecular mechanisms, we are using completely and semi-reconstituted fusion assays (Figure 2). These assays allow us to monitor content and lipid mixing and to detect hemifusion intermediates. In addition, we measure regulated exocytosis in vivo by using a pH-sensitive GFP (pHluorin) targeted to secretory vesicles. Cryo-electron microscopy will be employed to analyze the architecture of reconstituted fusion pores.
Specific projects: Role of lipids and membrane microdomains in exo- and endocytosis
t-SNAREs have been found in membrane microdomains and a perturbation of this microdomains inhibits exocytosis. Furthermore, the local lipid environment at the fusion site could significantly affect the recruitment of regulatory components, the membrane curvature, and fusion pore dynamics. Indeed recent results indicate that a local lipid turnover is required for exocytosis. Therefore we are analyzing the role of protein-lipid interactions and distinct lipids at different steps of the fusion reaction. Furthermore we will test, whether the unique lipid composition of synaptic vesicles facilitates the sorting of synaptic vesicle components and thus provides a platform for efficient endocytosis.
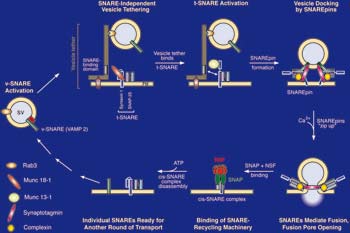 |
| Fig 1 . Model showing steps and components involved in neuronal exocytosis. |
Role of regulatory proteins (Rim1, Rab 3, Munc18, Munc13, complexins, synaptotagmins, Vo-ATPase, and other regulators)
Numerous regulators directly bind SNARE proteins, but in many cases their molecular functions are unclear and the regulatory network is poorly understood. We study these proteins as individual components and in various combinations. Therefore, these regulators are expressed as recombinant proteins in E.coli, or insect cells purified to homgeneity, and analyzed in in vitro fusion assays. In biochemical assays, we measure the fusion of an entire population of reconstiuted SNARE-liposomes (Figure 2A). In imaging assays, the docking, hemifusion, and full fusion of individual SNARE-lipodomes can be resolved (Figure 2B). In in vivo assays, the exocytosis of individual secretory vesicles is studied in living cells (Figure 2C). In this method, the secretory granules contain a pH-sensitive marker (pHluorin) in their lumen. Upon triggering exocyotosis, fusion pore opening and content release is detected by an increas in fluorescence. Using siRNA technology, proteins involved in exocytosis can be down regulated and replace by mutant copies. Thus, the biochemical results obtained in the reconstituted assays can be directly tested and verified under physiological conditions. These combined analyses shall reveal the sequential order and the entire assembly line that generates “ready to fuse” vesicles.
Structural and functional organization of fusion pores
At the electrophysiological level, fusion pores are well defined, but their structural organisation is unclear. The factors that determine reversible fusion pore opening/closure or full dilation are largely unknown. Thus we are attempting a structural analysis of reconstituted fusion intermediates by cryo-electronmicroscopy.
|
Fig. 2. In vitro and in vivo assays (A) Biochemical fusion assay. v- and t-SNAREs are reconstituted into small unilamellar donor and acceptor liposomes. Instead of acceptor liposomes, synaptic vesicles can be used. Membrane fusion of labeled donor liposomes with unlabeled acceptor liposomes results in fluorescence dequenching. |
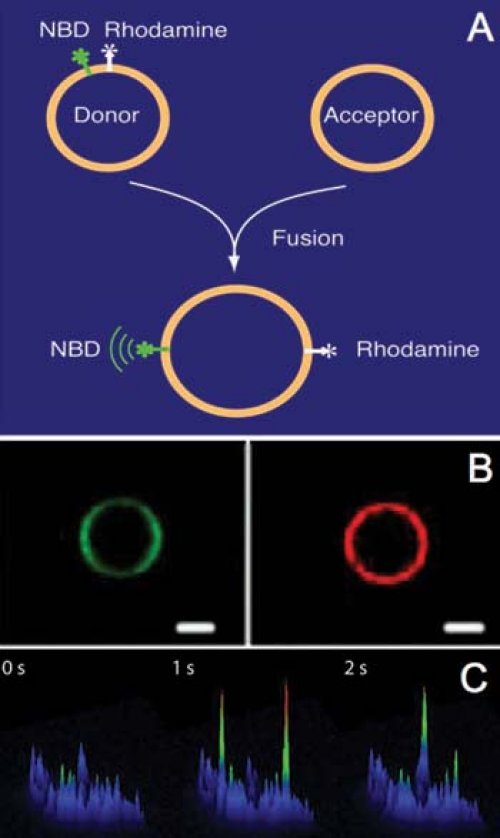 |
| (B) Single vesicle docking/fusion assay. A giant unilamellar vesicle, containing v-SNAREs is labeled with DiO-C18 (left panel). Addition of small unilamellar v-SNARE liposomes labeled with rhodamine PE shows vesicle docking (right panel). Scale bar 10 μM. | |
| (C) In vivo exocytosis assay. Fluorescence surface plots of a PC12 cell transfected with pHluorin targeted to secretory vesicles are shown. Membrane depolarization results in a transient increase of pHluorin fluorescence at single vesicle fusion sites (compare left panel with 2 right panels, s=seconds). |